2013年11月14日
结构生物学家,第一步确定蛋白质的精确的分子结构往往是最难的,哄骗蛋白质成长为有序,三维晶体大多数结构研究的起始物料。对特别困难的情况下,可能需要数年时间才能生成可用的晶体——有时蛋白质从未结晶尽管激烈的努力。霍华德休斯医学研究所日前科学家开发出一种新方法生成高分辨率的蛋白质结构的晶体小一百万倍比所需的x射线晶体学,最常见的方法确定蛋白质结构。
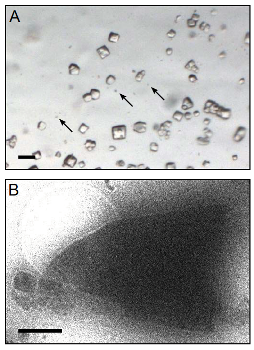 溶菌酶microrystals的法师。(一)光显微照片显示524溶菌酶微晶核(三个例子所示箭头)与525年相比大的晶体的大小通常用于x射线晶体学。比例尺是50 526米个人混合泳。(B)溶菌酶微晶核可视化在关注衍射模式收益而无视527年数据收集前低温电子显微镜。晶体的长度和宽度变化从528到6我估计厚度0.5 ~ 1我。比例尺是1 im。(Tamir Gonen, Janelia Farm Research Campus/HHMI)
溶菌酶microrystals的法师。(一)光显微照片显示524溶菌酶微晶核(三个例子所示箭头)与525年相比大的晶体的大小通常用于x射线晶体学。比例尺是50 526米个人混合泳。(B)溶菌酶微晶核可视化在关注衍射模式收益而无视527年数据收集前低温电子显微镜。晶体的长度和宽度变化从528到6我估计厚度0.5 ~ 1我。比例尺是1 im。(Tamir Gonen, Janelia Farm Research Campus/HHMI)
这项新技术,称为微,有可能加速结构生物学家的努力和扩大的蛋白质的高分辨率结构可以解决。“生化反应,它总是容易产生较小的晶体,“结构生物学家Gonen米尔说,他和同事们一起开发了技术在他的实验室里其本人是该园在霍华德·休斯医学研究所的研究校园。“有许多蛋白质,你不会得到晶体或结晶非常,非常小。他们可能会为微足够好。”
Gonen和他的同事描述微在11月19日发表的一份报告,2013年开放获取期刊eLife。使用Gonen的新技术,科学家可以利用电子从电子显微镜来确定蛋白质微晶核的结构。这种方法被称为电子晶体学,之前仅限于研究的蛋白质,可以成长为很薄,二维晶体。“人们把3 d晶体在电子显微镜,但没有人能够解决他们的结构,“Gonen说。
这是因为从显微镜的高能电子束辐射——这是由蛋白质晶体分散,创建衍射模式,研究人员使用推断结构——迅速破坏了蛋白质晶体。水晶生存两个或三个曝光“低剂量”光束,Gonen解释道。然而,许多需要更多的衍射模式来揭示一个蛋白质的结构。
这意味着生成足够的数据来生成一个结构,研究人员必须收集和分析来自成千上万的不同晶体的衍射模式。结合这些数据是很困难的甚至是二维晶体,但对于三维晶体,这证明了一个不可逾越的任务。“如果数据来自成千上万的不同晶体,你不知道你在哪里在倒易空间——不知道这些不同的衍射模式之间的关系。没有办法指数和确定结构的一切,”Gonen说。
所以Gonen,显微镜专家丹·施和他们的同事们决定简化数据处理通过收集许多从单晶衍射模式。“当我们收集多个衍射模式从单一晶体,它们之间的连续性。我们知道它们是如何联系的,”他解释说。
“我们不知道如果我们可以收集整个数据集的单晶,但我们决定尝试,”他说。团队知道保留水晶重复曝光,他们将不得不大幅减少电子显微镜的剂量。“大多数人使用电子显微镜成像,”Gonen解释道。“但是我们只是想收集衍射模式以来,我们能够使用200倍低于通常被认为是低剂量”。
为了测试他们的想法,布伦特Nannenga Gonen的实验室的一位博士后研究员,非常小的蛋白溶菌酶晶体增长,一个已知的结构。他们应用这些微晶核,如此之小的出现仅仅是斑点在光学显微镜下,一个小碳网格。将网格到显微镜后,他们编程工具,这样它将旋转网格1度,每次旋转后收集衍射图案。九十-价值度的数据收集。
马特•Iadanza Gonen研究生的实验室,开发的软件索引90衍射模式和集成,使团队来生成一个完整的溶菌酶蛋白的结构。提高结构的质量,他们重复这个过程两个晶体——每个躺在一个不同的方向的网格,然后结合数据到一个单独的结构。
结构符合已知的溶菌酶的结构,分辨率为2.9埃。“这正是你想要的生物蛋白,“Gonen说,解释说,这种级别的细节入手的许多结构图案是蛋白质功能的基础。“决议,你真的可以开始理解分子机制,”他说。通过进一步细化他们的数据处理程序,他补充说,他的团队应该能够提高微提供的分辨率。数据已经收集到1.7埃分辨率,所以这只是一个时间问题。
Gonen的团队目前正在与安德鲁。莱斯利医学研究委员会分子生物学实验室将微数据处理集成到标准的x射线晶体学软件(MOSFLM),使技术更容易与其他实验室。与此同时,他说,许多实验室已经把蛋白质微晶核Janelia,和团队正在调查技术解决未知结构的潜力。
”有很多的兴奋,“Gonen说,“因为很多人微晶核,他们甚至不知道它。我们发现有大量晶体的结晶下降总是微晶核。即使你看不到晶体在光学显微镜下,这并不意味着你没有微晶核。”
新闻稿avilable从http://www.newswise.com/
来源:http://www.hhmi.org