介绍gydF4y2Ba近年来,它已被广泛证明紫外线辐射和之间的交互gydF4y2Ba半导体催化剂gydF4y2Ba有很强的工业废水中有毒有机物的破坏潜力gydF4y2Ba(gydF4y2Ba1、2、3gydF4y2Ba]gydF4y2Ba。光催化有许多潜在的应用治疗包含有机物的水,从水中去除金属和分裂水。gydF4y2Ba所有这些过程都是相互关联的。gydF4y2Ba带隙半导体粒子悬浮在水中的照明使电子从价带过渡到传导带产生电子空穴对。gydF4y2Ba同时,在流体相的存在,发生吸附和电子转移收益对受体分子(氧化物种),而积极的洞是转移到供体分子gydF4y2Ba(gydF4y2Ba4gydF4y2Ba]gydF4y2Ba。gydF4y2Ba 当前催化研究的一个主要中心的关注是关键结构关系的说明现有的固体催化剂,可以随后用于材料的设计。亚博网站下载这些研究需要的组合材料合成、表征和催化测试。亚博网站下载gydF4y2Ba 井上et al .,表明钠hexatitanate NagydF4y2Ba2gydF4y2Ba“透明国际”gydF4y2Ba6gydF4y2BaOgydF4y2Ba13gydF4y2Ba如俄罗斯,加上一个活跃的阶段,有能力的调用一个光催化分解水产生HgydF4y2Ba2gydF4y2Ba和OgydF4y2Ba2gydF4y2Ba大约在stoichometric比例gydF4y2Ba(gydF4y2Ba5gydF4y2Ba]gydF4y2Ba。gydF4y2Ba这种混合氧化物是一种碱金属钛属于M的化学公式gydF4y2Ba2gydF4y2Ba“透明国际”gydF4y2BangydF4y2BaOgydF4y2Ba2 n + 1gydF4y2Ba(碱金属原子)。gydF4y2Ba 另一方面,以往的研究对三元BaO-Li阶段形成的系统gydF4y2Ba2gydF4y2BaO-TiOgydF4y2Ba2gydF4y2Ba的成分包含> TiO的50%gydF4y2Ba2gydF4y2Ba,gydF4y2Ba证明了阶段,集中在英航gydF4y2Ba3gydF4y2Ba李gydF4y2Ba2gydF4y2Ba“透明国际”gydF4y2Ba8gydF4y2BaOgydF4y2Ba20.gydF4y2Ba显示,电子导电率gydF4y2Ba(gydF4y2Ba6、7gydF4y2Ba]gydF4y2Ba,这表明gydF4y2Ba这种化合物可以用作半导体。的晶体结构gydF4y2Ba英航gydF4y2Ba3gydF4y2Ba李gydF4y2Ba2gydF4y2Ba“透明国际”gydF4y2Ba8gydF4y2BaOgydF4y2Ba20.gydF4y2Ba类似于Na的吗gydF4y2Ba2gydF4y2Ba“透明国际”gydF4y2Ba6gydF4y2BaOgydF4y2Ba13gydF4y2Ba和gydF4y2Ba英航gydF4y2Ba2gydF4y2Ba“透明国际”gydF4y2Ba6gydF4y2BaOgydF4y2Ba13gydF4y2Ba(gydF4y2Ba单斜晶体的空间群C2 / m(12路)gydF4y2Ba]gydF4y2Ba在gydF4y2Ba这edge-sharinggydF4y2Ba正八面体的三聚形成的三种类型:Ti (1) OgydF4y2Ba6gydF4y2Ba李,Ti / (2) OgydF4y2Ba6gydF4y2Ba和Ti (3) OgydF4y2Ba6gydF4y2Ba,与其他三聚形成共享角落无限丝带与曲折的安排。gydF4y2Ba隧道内的英航原子之间形成的丝带edge-sharing正八面体gydF4y2Ba(gydF4y2Ba8gydF4y2Ba]gydF4y2Ba。gydF4y2Ba 我们假设如果催化剂的结构类似,钛酸三元英航gydF4y2Ba3gydF4y2Ba李gydF4y2Ba2gydF4y2Ba“透明国际”gydF4y2Ba8gydF4y2BaOgydF4y2Ba20.gydF4y2Ba或其他钛具有相同的结构,也应该表现出光催化行为。gydF4y2Ba调查这个假设我们2的分解反应进行,4 dinitroaniline除以相同的降解条件下获得的固体。gydF4y2Ba然而,在光催化半导体催化活性的不仅是由电子性质决定的,也是由粒子大小、表面积和结晶度。gydF4y2Ba 样品的化学历史和热处理gydF4y2Ba非常重要的gydF4y2Ba活动的半导体gydF4y2Ba(gydF4y2Ba9-14gydF4y2Ba]gydF4y2Ba;因此,在这项工作中,我们准备了陶瓷化合物gydF4y2Ba2gydF4y2Ba“透明国际”gydF4y2Ba6gydF4y2BaOgydF4y2Ba13gydF4y2Ba,NagydF4y2Ba2gydF4y2Ba“透明国际”gydF4y2Ba6gydF4y2BaOgydF4y2Ba13gydF4y2Ba和KgydF4y2Ba2gydF4y2Ba“透明国际”gydF4y2Ba6gydF4y2BaOgydF4y2Ba13gydF4y2Ba通过溶胶-凝胶法和固态反应为了研究它们的光催化特性和制备方法的影响。gydF4y2Ba英航的gydF4y2Ba3gydF4y2Ba李gydF4y2Ba2gydF4y2Ba“透明国际”gydF4y2Ba8gydF4y2BaOgydF4y2Ba20.gydF4y2Ba只是由溶胶-凝胶技术在酸性和碱性条件。gydF4y2Ba 实验gydF4y2Ba合成gydF4y2Ba为gydF4y2Ba一个gydF4y2Ba米gydF4y2Ba米gydF4y2Ba2 ngydF4y2BaOgydF4y2Ba4 n + 1gydF4y2Ba(A = Li Na, K;m = 2, n = 3)gydF4y2Ba溶胶-凝胶法合成,我们使用金属前体钛n-butoxide (98 + %, Strem化学)和相应的碱金属醋酸(99.5%,奥尔德里奇),t-butanol (99.5%, j·t·贝克)用作溶剂和冰醋酸(奥德里奇99%)添加在连续搅拌达到pH值3。gydF4y2Ba 为gydF4y2Ba英航gydF4y2Ba3gydF4y2Ba李gydF4y2Ba2gydF4y2Ba“透明国际”gydF4y2Ba8gydF4y2BaOgydF4y2Ba20.gydF4y2Ba溶胶-凝胶法,gydF4y2Baarium异丙醇盐(奥德里奇化工99.9%),醋酸锂二水合物(σ63.4%)和钛异丙醇盐(奥德里奇化工97%)是金属前体。gydF4y2Ba乙醇(99.9% PROQUIM)被用作溶剂和冰醋酸(PROQUIM 99.8%)添加在连续搅拌下达到pH值3gydF4y2Ba(gydF4y2Ba15gydF4y2Ba]gydF4y2Ba。gydF4y2Ba反应混合物加热回流在70°C。gydF4y2Ba在空气干燥获得的凝胶在80°C 48 h(新鲜样品)和不同部分被加热在200,400、600和800°CgydF4y2Ba6 h。gydF4y2Ba这些三元化合物gydF4y2Ba也准备在9通过溶胶-凝胶法和pH值gydF4y2Ba氢氧化氨代替醋酸gydF4y2Ba(gydF4y2Ba16gydF4y2Ba]gydF4y2Ba。gydF4y2Ba 只有二元化合物gydF4y2Ba合成了固态反应。gydF4y2Ba一种亲密TiO的混合物gydF4y2Ba2gydF4y2Ba和相应的碳酸盐gydF4y2Ba(奥尔德里奇99%)被加热在1250ºC在48 - 72 h。gydF4y2Ba 描述gydF4y2Ba红外吸收光谱是珀金埃尔默gydF4y2Ba傅里叶变换我gydF4y2Banfrared光谱学gydF4y2Ba(gydF4y2Ba傅立叶变换红外光谱gydF4y2Ba)gydF4y2Ba1000年光谱仪典范gydF4y2Ba电脑为了分析催化剂上的羟基。gydF4y2Ba氮吸附等温线测定样品的气体吸附仪(尽快微粒学2000)。gydF4y2Ba比表面积是由BET方程计算。gydF4y2Ba分析的样本gydF4y2Bax射线衍射gydF4y2Ba(XRD)gydF4y2Ba技术gydF4y2Ba在西门子d - 5000与铜衍射仪gydF4y2BaKαgydF4y2Ba辐射gydF4y2Ba(λ=gydF4y2Ba1.5418gydF4y2Ba)。gydF4y2Ba固体的漫反射光谱得到的珀金埃尔默分光光度计λ12耦合集成Labsphere RSA-PE-20。gydF4y2BaSpectralon标准usr - 99010反射率为100%被用作参考。gydF4y2Ba 光催化gydF4y2Ba活动gydF4y2BaDgydF4y2BainitroanilinegydF4y2BaDgydF4y2BaecompositiongydF4y2Ba光催化实验在室温下进行,200毫克的每个催化剂添加到一个包含500毫升瓶的水溶液中2,4-dinitroaniline 30 ppm。gydF4y2Ba在搅拌下,解决方案与紫外线灯辐照在一个封闭的盒子XX-15 Black-Ray模型,它发出的辐射gydF4y2BaλgydF4y2Ba1100 = 254 nm的强度gydF4y2BaμgydF4y2BaW /厘米gydF4y2Ba2gydF4y2Ba。gydF4y2Ba反应速率又以每10分钟,然后分析了整除gydF4y2BangydF4y2Ba紫外可见分光光度计gydF4y2Ba优秀的Lambda-12gydF4y2Ba。gydF4y2Ba2的浓度,计算4-dinitroaniline吸收带的346海里出现在这种物质的紫外可见光谱。gydF4y2Ba数据评估我们应用Langmuir-Hinshelwood模型,使用多相光催化反应方程的一批反应堆gydF4y2Ba(gydF4y2Ba17gydF4y2Ba]gydF4y2Ba。gydF4y2Ba 估计中C =有限公司/ 2的时候,我们使用:gydF4y2Ba 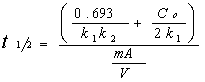
C V是液体的体积,底物浓度,gydF4y2BatgydF4y2Ba时间,gydF4y2Ba米gydF4y2Ba催化剂的质量,g /猫的吸附位点,gydF4y2BakgydF4y2Ba1gydF4y2Ba表观速率常数gydF4y2BakgydF4y2Ba2gydF4y2Ba明显的吸附常数。gydF4y2Ba 减少铬六世gydF4y2Ba钠的结晶形式gydF4y2Ba2gydF4y2Ba“透明国际”gydF4y2Ba6gydF4y2BaOgydF4y2Ba13gydF4y2Ba和英航gydF4y2Ba3gydF4y2Ba李gydF4y2Ba2gydF4y2Ba“透明国际”gydF4y2Ba8gydF4y2BaOgydF4y2Ba20.gydF4y2Ba和陶瓷化合物gydF4y2BaNagydF4y2Ba2gydF4y2BaZrTigydF4y2Ba5gydF4y2BaOgydF4y2Ba13gydF4y2Ba和钠gydF4y2Ba1.8gydF4y2Ba英航gydF4y2Ba0.3gydF4y2Ba“透明国际”gydF4y2Ba5.9gydF4y2BaOgydF4y2Ba13gydF4y2Ba由溶胶-凝胶法,gydF4y2Ba被用作催化剂photacatalytic减少铬(VI)的铬(III)。gydF4y2Ba在这些实验中100 - 15所示gydF4y2Ba0毫克的每个催化剂添加到一个包含200毫升瓶的水溶液中KgydF4y2Ba2gydF4y2BaCrgydF4y2Ba2gydF4y2BaOgydF4y2Ba7gydF4y2Ba(试剂级)30 ppm。gydF4y2Ba解决方案的初始pH值是1 - 2。gydF4y2Ba液体维持在连续搅拌时辐照在封闭框相同的紫外灯用于2、4 dinitroaniline退化。gydF4y2Ba分析了铬(VI)的浓度使用diphenylcarbazide紫外光谱gydF4y2Ba(gydF4y2Ba18gydF4y2Ba]gydF4y2Ba。gydF4y2Ba 所有实验,开始解决方案与催化剂被允许平衡没有紫外线光照30分钟。gydF4y2Ba 结果gydF4y2Ba和讨论gydF4y2BaX射线衍射gydF4y2Ba图1显示了XRD的钛酸锂的干凝胶演化模式与温度。gydF4y2Ba当粉加热在200和400°C几个山峰是观察到,这是表明几乎非晶态结构。gydF4y2Ba然而,一个小峰是观察到25°相关TiO的主峰gydF4y2Ba2gydF4y2Ba锐钛矿相。gydF4y2Ba当新鲜样品加热在800°C,干凝胶转化为产生晶体材料粉末模式类似报道gydF4y2Ba李gydF4y2Ba2gydF4y2Ba“透明国际”gydF4y2Ba6gydF4y2BaOgydF4y2Ba13。gydF4y2Ba固态法,根据XRD研究,是不可能获得这种化合物,这是由于一些锂在高温挥发损失。gydF4y2Ba

|
无花果gydF4y2Ba保证1。gydF4y2BaXRDgydF4y2Ba溶胶-凝胶法钛酸锂的热演化模式gydF4y2Ba。gydF4y2Ba |
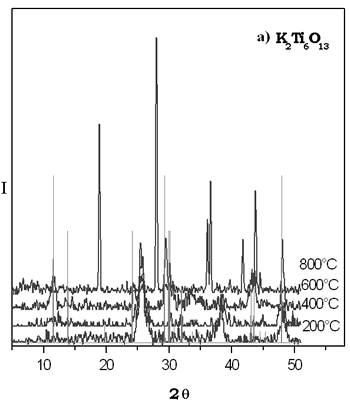
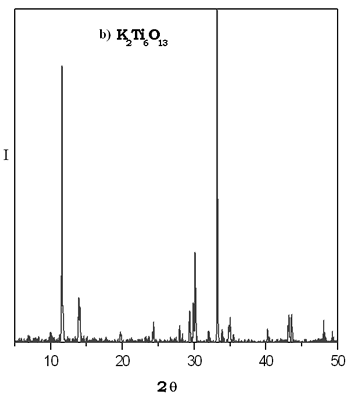
Na的形成gydF4y2Ba2gydF4y2Ba“透明国际”gydF4y2Ba6gydF4y2BaOgydF4y2Ba13gydF4y2Ba实现复合溶胶-凝胶法获得的样本加热时也在800°C。gydF4y2Ba在600ºC锐钛矿的形式存在。gydF4y2Ba通过陶瓷的方法,这种化合物形成在1250°C 48 h。gydF4y2Ba  XRD干凝胶的热演化模式的钛酸钾如图2所示。gydF4y2Ba我们可以看到,最重要的反映复合KgydF4y2Ba2gydF4y2Ba“透明国际”gydF4y2Ba6gydF4y2BaOgydF4y2Ba13gydF4y2Ba被安装在600°C。gydF4y2Ba小反射指出hexatitanate钾的存在形成了纳米晶体的粒度区间。gydF4y2Ba很明显在样本加热在800°C,反思观察不对应于KgydF4y2Ba2gydF4y2Ba“透明国际”gydF4y2Ba6gydF4y2BaOgydF4y2Ba13gydF4y2Ba结构。gydF4y2Ba这对于TiO difractogram类似于报道gydF4y2Ba2gydF4y2Ba金红石形成稳定在较高的温度,这是可能的溶胶-凝胶相是亚稳态和600ºC成为其他水晶硬币,然后大多数阶段在800°C的粉是金红石。gydF4y2Ba通过固态法KgydF4y2Ba2gydF4y2Ba“透明国际”gydF4y2Ba6gydF4y2BaOgydF4y2Ba13gydF4y2Ba化合物在1250°C到72年获得h。gydF4y2Ba XRD干凝胶的热演化模式的钛酸钾如图2所示。gydF4y2Ba我们可以看到,最重要的反映复合KgydF4y2Ba2gydF4y2Ba“透明国际”gydF4y2Ba6gydF4y2BaOgydF4y2Ba13gydF4y2Ba被安装在600°C。gydF4y2Ba小反射指出hexatitanate钾的存在形成了纳米晶体的粒度区间。gydF4y2Ba很明显在样本加热在800°C,反思观察不对应于KgydF4y2Ba2gydF4y2Ba“透明国际”gydF4y2Ba6gydF4y2BaOgydF4y2Ba13gydF4y2Ba结构。gydF4y2Ba这对于TiO difractogram类似于报道gydF4y2Ba2gydF4y2Ba金红石形成稳定在较高的温度,这是可能的溶胶-凝胶相是亚稳态和600ºC成为其他水晶硬币,然后大多数阶段在800°C的粉是金红石。gydF4y2Ba通过固态法KgydF4y2Ba2gydF4y2Ba“透明国际”gydF4y2Ba6gydF4y2BaOgydF4y2Ba13gydF4y2Ba化合物在1250°C到72年获得h。gydF4y2Ba
|
图2。gydF4y2Ba一)gydF4y2BaXRDgydF4y2Ba钛酸钾凝胶的热演化模式gydF4y2Ba。b)gydF4y2BaXRDgydF4y2Ba的模式gydF4y2BaKgydF4y2Ba2gydF4y2Ba“透明国际”gydF4y2Ba6gydF4y2BaOgydF4y2Ba13gydF4y2Ba由固态反应gydF4y2Ba。gydF4y2Ba |
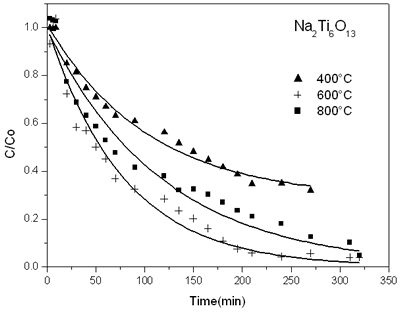
英航的形成gydF4y2Ba3gydF4y2Ba李gydF4y2Ba2gydF4y2Ba“透明国际”gydF4y2Ba8gydF4y2BaOgydF4y2Ba20.gydF4y2Ba溶胶-凝胶法实现了pH值3当一个新鲜样本加热在800°C。gydF4y2Ba晶体结构是由XRD确认模式类似报道Dussarrat和et al。gydF4y2Ba(gydF4y2Ba8gydF4y2Ba]gydF4y2Ba。gydF4y2Ba 当一个三元化合物合成了由溶胶凝胶在pH值9日里特维德研究表明BagydF4y2Ba3gydF4y2Ba李gydF4y2Ba2gydF4y2Ba“透明国际”gydF4y2Ba8gydF4y2BaOgydF4y2Ba20.gydF4y2Ba形成的纳米晶体的粒度区间在600°CgydF4y2Ba(gydF4y2Ba19gydF4y2Ba]gydF4y2Ba。gydF4y2BaNanocrystallinity发起的化合物在较低温度时,溶胶-凝胶法在碱性博士样品退火在800°C显示x射线衍射模式对应于英航在文献中报道gydF4y2Ba3gydF4y2Ba李gydF4y2Ba2gydF4y2Ba“透明国际”gydF4y2Ba8gydF4y2BaOgydF4y2Ba20.gydF4y2Ba结构,但它有更高的结晶度。gydF4y2Ba 光催化活性gydF4y2BaDinitroaniline分解gydF4y2Ba图3显示了tgydF4y2Ba他减少2、4 dinitroaniline浓度和反应时间对钛酸钠催化剂由溶胶-凝胶法。gydF4y2Ba相似的图形和其他固体做准备。gydF4y2Ba我们可以看到,样品越活跃在600ºC时获得的固体NagydF4y2Ba2gydF4y2Ba“透明国际”gydF4y2Ba6gydF4y2BaOgydF4y2Ba13gydF4y2Ba结构形成。gydF4y2Ba
|
图3。gydF4y2Ba进化的时间函数分解的2、4 dinitroaniline在钛酸钠催化剂溶胶-凝胶法在不同温度退火。gydF4y2Ba |
的值gydF4y2BakgydF4y2Ba1gydF4y2BakgydF4y2Ba2gydF4y2BakgydF4y2Ba1gydF4y2BakgydF4y2Ba2gydF4y2Ba(明显反应)和tgydF4y2Ba1/2gydF4y2Ba(明显的半衰期)对于每一个催化剂,2的分解4-dinitroaniline,gydF4y2Ba应用Langmuir-Hinshelwood计算模型。gydF4y2Ba这些数据都是估计的图像(Ln(公司/ C)) / (Co-C)gydF4y2Bat / (Co-C)。gydF4y2Ba 每个半导体催化剂的光催化活性,因此,相比使用半衰期时间值如表1所示。gydF4y2Ba 表1。gydF4y2Ba半衰期时间值的2.4 -dinitroaniline的分解gydF4y2Ba一个gydF4y2Ba米gydF4y2Ba米gydF4y2Ba2 ngydF4y2BaOgydF4y2Ba4 ngydF4y2Ba+ 1gydF4y2Ba溶胶-凝胶催化剂gydF4y2Ba。gydF4y2Ba
|
李gydF4y2Ba2gydF4y2Ba“透明国际”gydF4y2Ba6gydF4y2BaOgydF4y2Ba13gydF4y2Ba |
400年gydF4y2Ba |
3.13gydF4y2Ba |
93.75gydF4y2Ba |
34gydF4y2Ba |
李gydF4y2Ba2gydF4y2Ba“透明国际”gydF4y2Ba6gydF4y2BaOgydF4y2Ba13gydF4y2Ba |
600年gydF4y2Ba |
3.19gydF4y2Ba |
17.59gydF4y2Ba |
57gydF4y2Ba |
李gydF4y2Ba2gydF4y2Ba“透明国际”gydF4y2Ba6gydF4y2BaOgydF4y2Ba13gydF4y2Ba |
800年gydF4y2Ba |
3.39gydF4y2Ba |
0.19gydF4y2Ba |
25gydF4y2Ba |
NagydF4y2Ba2gydF4y2Ba“透明国际”gydF4y2Ba6gydF4y2BaOgydF4y2Ba13gydF4y2Ba |
400年gydF4y2Ba |
3.15gydF4y2Ba |
73.1gydF4y2Ba |
297年gydF4y2Ba |
NagydF4y2Ba2gydF4y2Ba“透明国际”gydF4y2Ba6gydF4y2BaOgydF4y2Ba13gydF4y2Ba |
600年gydF4y2Ba |
3.17gydF4y2Ba |
60.7gydF4y2Ba |
67年gydF4y2Ba |
NagydF4y2Ba2gydF4y2Ba“透明国际”gydF4y2Ba6gydF4y2BaOgydF4y2Ba13gydF4y2Ba |
800年gydF4y2Ba |
3.18gydF4y2Ba |
7.69gydF4y2Ba |
120年gydF4y2Ba |
KgydF4y2Ba2gydF4y2Ba“透明国际”gydF4y2Ba6gydF4y2BaOgydF4y2Ba13gydF4y2Ba |
400年gydF4y2Ba |
3.14gydF4y2Ba |
- - - - -gydF4y2Ba |
131年gydF4y2Ba |
KgydF4y2Ba2gydF4y2Ba“透明国际”gydF4y2Ba6gydF4y2BaOgydF4y2Ba13gydF4y2Ba |
600年gydF4y2Ba |
3.19gydF4y2Ba |
37.9gydF4y2Ba |
91年gydF4y2Ba |
KgydF4y2Ba2gydF4y2Ba“透明国际”gydF4y2Ba6gydF4y2BaOgydF4y2Ba13gydF4y2Ba*gydF4y2Ba |
800年gydF4y2Ba |
3.46gydF4y2Ba |
0.51gydF4y2Ba |
103年gydF4y2Ba |
* TiOgydF4y2Ba2gydF4y2Ba大多数结晶相金红石水的形式存在gydF4y2Ba 与溶胶-凝胶催化剂,我们可以观察到样品,锂是最活跃的催化剂材料加热在800ºC时锂hexatitanate已经形成的结构。gydF4y2Ba在溶胶-凝胶法钠样品的情况下,最好的活动表现出了固体加热TiO时在600ºCgydF4y2Ba2gydF4y2Ba锐钛矿形式存在。gydF4y2Ba以前在TiO的工作gydF4y2Ba2gydF4y2Ba溶胶-凝胶法合成gydF4y2Ba(gydF4y2Ba20.gydF4y2Ba]gydF4y2BapH3条件表明,锐钛矿存在小百分比的金红石样品退火温度较低时(200 - 600ºC)。gydF4y2Ba一些调查在光催化降解不同的有机化合物在水里,表明积极的锐钛矿形式比金红石相gydF4y2Ba(gydF4y2Ba21日,gydF4y2Ba22gydF4y2Ba]gydF4y2Ba。gydF4y2Ba在这种情况下,结构是由锐钛矿NagydF4y2Ba2gydF4y2Ba“透明国际”gydF4y2Ba6gydF4y2BaOgydF4y2Ba13gydF4y2Ba在800ºC。gydF4y2Ba 钾材料,活动是改善催化剂加热亚博网站下载在600ºC。gydF4y2Ba我们已经看到在这个温度gydF4y2BaKgydF4y2Ba2gydF4y2Ba“透明国际”gydF4y2Ba6gydF4y2BaOgydF4y2Ba13gydF4y2Ba结构是实现小粒径青睐的光催化活性。gydF4y2Ba在800ºC,金红石形式存在和几个工作报告较低的活动的催化剂。空穴和电子复合的速率较高的金红石与锐钛矿由于两个TiO的半导体性质gydF4y2Ba2gydF4y2Ba不同的阶段gydF4y2Ba(gydF4y2Ba23gydF4y2Ba]gydF4y2Ba。gydF4y2Ba 在所有的溶胶-凝胶法gydF4y2Ba一个gydF4y2Ba米gydF4y2Ba米gydF4y2Ba2 ngydF4y2BaOgydF4y2Ba4 n + 1gydF4y2Ba催化剂探索我们可以看到最好的活动是通过锂hexatitanate获得在800ºC。gydF4y2BaTiO的gydF4y2Ba6gydF4y2Ba正八面体的畸变是由于碱性原子的大小。gydF4y2Ba这些实验不扭曲的结构表现出的光催化降解的主要活动2,4-dinitroaniline。gydF4y2Ba 另一方面,根据与半衰期时间计算催化剂由陶瓷方法(表2),我们可以欣赏到一个伟大的催化活性类似于锂hexatitanate溶胶-凝胶法。gydF4y2Ba在这些材料结晶度更亚博网站下载高和带隙energgydF4y2BaygydF4y2Ba值计算的紫外可见光谱分别为3.04和3.08 eVgydF4y2BaNagydF4y2Ba2gydF4y2Ba“透明国际”gydF4y2Ba6gydF4y2BaOgydF4y2Ba13gydF4y2Ba和gydF4y2BaKgydF4y2Ba2gydF4y2Ba“透明国际”gydF4y2Ba6gydF4y2BaOgydF4y2Ba13gydF4y2Ba分别,而对于相同的化合物通过溶胶-凝胶法得到这个值是3.4 eVgydF4y2Ba李gydF4y2Ba2gydF4y2Ba“透明国际”gydF4y2Ba6gydF4y2BaOgydF4y2Ba13gydF4y2Ba和gydF4y2BaNagydF4y2Ba2gydF4y2Ba“透明国际”gydF4y2Ba6gydF4y2BaOgydF4y2Ba13gydF4y2Ba;gydF4y2Ba不过gydF4y2Ba为gydF4y2BaKgydF4y2Ba2gydF4y2Ba“透明国际”gydF4y2Ba6gydF4y2BaOgydF4y2Ba13gydF4y2Ba是3.1 ev。gydF4y2Ba 表2gydF4y2Ba。gydF4y2Ba半衰期时间值的2.4 -dinitroaniline的分解gydF4y2Ba一个gydF4y2Ba米gydF4y2Ba米gydF4y2Ba2 ngydF4y2BaOgydF4y2Ba4 ngydF4y2Ba+ 1gydF4y2Ba催化剂由固态反应gydF4y2Ba。gydF4y2Ba
|
NagydF4y2Ba2gydF4y2Ba“透明国际”gydF4y2Ba6gydF4y2BaOgydF4y2Ba13gydF4y2Ba |
1250年gydF4y2Ba |
3.08gydF4y2Ba |
0.4gydF4y2Ba |
21gydF4y2Ba |
KgydF4y2Ba2gydF4y2Ba“透明国际”gydF4y2Ba6gydF4y2BaOgydF4y2Ba13gydF4y2Ba |
1250年gydF4y2Ba |
3.04gydF4y2Ba |
0.4gydF4y2Ba |
16gydF4y2Ba |
一旦英航gydF4y2Ba3gydF4y2Ba李gydF4y2Ba2gydF4y2Ba“透明国际”gydF4y2Ba8gydF4y2BaOgydF4y2Ba20.gydF4y2Ba作为光催化剂,我们发现更高的光催化活性表达为tgydF4y2Ba1/2gydF4y2Ba(表3),gydF4y2Ba在pH值3和获得的固体退火在800°C (tgydF4y2Ba1/2gydF4y2Ba=gydF4y2Ba26分钟)。gydF4y2Ba这种催化剂的吸附容量青睐是因为它羟基化程度更高,因此哦组表现为吸附中心。gydF4y2Ba在这个温度(800°C),英航的晶体结构gydF4y2Ba3gydF4y2Ba李gydF4y2Ba2gydF4y2Ba“透明国际”gydF4y2Ba8gydF4y2BaOgydF4y2Ba20.gydF4y2Ba根据XRD研究形成gydF4y2Ba。gydF4y2Ba 表3。gydF4y2Ba半衰期时间值的2.4 -dinitroaniline的分解gydF4y2Ba英航gydF4y2Ba3gydF4y2Ba李gydF4y2Ba2gydF4y2Ba“透明国际”gydF4y2Ba8gydF4y2BaOgydF4y2Ba20.gydF4y2Ba溶胶-凝胶法的催化剂。gydF4y2Ba
|
3gydF4y2Ba |
400年gydF4y2Ba |
3.09gydF4y2Ba |
66.51gydF4y2Ba |
44gydF4y2Ba |
3gydF4y2Ba |
600年gydF4y2Ba |
3.14gydF4y2Ba |
38.08gydF4y2Ba |
33gydF4y2Ba |
3gydF4y2Ba |
800年gydF4y2Ba |
3.18gydF4y2Ba |
6.17gydF4y2Ba |
26gydF4y2Ba |
|
|
|
|
|
9gydF4y2Ba |
400年gydF4y2Ba |
3.48gydF4y2Ba |
117.83gydF4y2Ba |
54gydF4y2Ba |
9gydF4y2Ba |
600年gydF4y2Ba |
3.44gydF4y2Ba |
49.30gydF4y2Ba |
24gydF4y2Ba |
9gydF4y2Ba |
800年gydF4y2Ba |
3.37gydF4y2Ba |
9.37gydF4y2Ba |
28gydF4y2Ba |
的三元催化剂合成pH9样品发射的光降解提高600和800ºC。gydF4y2Ba在基本条件示例煅烧800ºC比在酸性条件下具有相同的晶体结构,然而,它失去了几乎所有导致羟基组gydF4y2Ba缺陷程度高gydF4y2Ba产生空缺。gydF4y2Ba氧化物半导体与电子缺陷的属性。gydF4y2Ba根据gydF4y2BakgydF4y2Ba1gydF4y2Ba值计算gydF4y2Ba,gydF4y2Ba在这个示例中,电子转移是青睐,它显示了一个好的活动2,4-dinitroaniline分解(tgydF4y2Ba1/2gydF4y2Ba= 28分钟)。gydF4y2Ba然而,吸附容量低是由于材料热处理烧结发生。gydF4y2Ba与样品加热在600ºC获得碱性pH值,该活动表示为tgydF4y2Ba1/2gydF4y2Ba= 24分钟,接近那些计算样品退火在800ºC。gydF4y2Ba这种行为主要是由于比表面积和ngydF4y2Baanocrynstallinity三元化合物。gydF4y2Ba 铬六世gydF4y2Ba减少gydF4y2Ba表4显示了光催化活性的初步研究的结果gydF4y2BaNagydF4y2Ba2gydF4y2Ba“透明国际”gydF4y2Ba6gydF4y2BaOgydF4y2Ba13gydF4y2Ba和英航gydF4y2Ba3gydF4y2Ba李gydF4y2Ba2gydF4y2Ba“透明国际”gydF4y2Ba8gydF4y2BaOgydF4y2Ba20.gydF4y2Ba与他人相比同构的化合物(gydF4y2BaNagydF4y2Ba2gydF4y2BaZrTigydF4y2Ba5gydF4y2BaOgydF4y2Ba13gydF4y2Ba和gydF4y2BaNagydF4y2Ba1.8gydF4y2Ba英航gydF4y2Ba0.3gydF4y2Ba“透明国际”gydF4y2Ba5.9gydF4y2BaOgydF4y2Ba13gydF4y2Ba)gydF4y2Ba六价铬光致还原作用。gydF4y2Ba活动评估计算的一部分铬(VI)减少到铬(III)后3.5 h(紫外线光照。gydF4y2Ba动力学研究是我们研究小组为了评估这些化合物的反应速率。gydF4y2Ba 表4gydF4y2Ba。gydF4y2Ba铬(VI)光致还原作用的同构的材料百分比与单斜结构空间群C2 / m亚博网站下载gydF4y2Ba。gydF4y2Ba
|
NagydF4y2Ba2gydF4y2Ba“透明国际”gydF4y2Ba6gydF4y2BaOgydF4y2Ba13gydF4y2Ba |
86年gydF4y2Ba |
15.13gydF4y2Ba |
3.74gydF4y2Ba |
9.16gydF4y2Ba |
99.30gydF4y2Ba |
英航gydF4y2Ba3gydF4y2Ba李gydF4y2Ba2gydF4y2Ba“透明国际”gydF4y2Ba8gydF4y2BaOgydF4y2Ba20.gydF4y2Ba |
71年gydF4y2Ba |
15.17gydF4y2Ba |
3.90gydF4y2Ba |
9.11gydF4y2Ba |
98.64gydF4y2Ba |
NagydF4y2Ba2gydF4y2BaZrTigydF4y2Ba5gydF4y2BaOgydF4y2Ba13gydF4y2Ba |
53gydF4y2Ba |
- - - - - -gydF4y2Ba |
- - - - - -gydF4y2Ba |
- - - - - -gydF4y2Ba |
- - - - - -gydF4y2Ba |
NagydF4y2Ba1.8gydF4y2Ba英航gydF4y2Ba0.3gydF4y2Ba“透明国际”gydF4y2Ba5.9gydF4y2BaOgydF4y2Ba13gydF4y2Ba |
31日gydF4y2Ba |
15.18gydF4y2Ba |
3.78gydF4y2Ba |
9.14gydF4y2Ba |
98.70gydF4y2Ba |
结论gydF4y2Ba当溶胶-凝胶方法被用来准备陶瓷粉,酸条件造成化合物的形成与减少表面积和孔隙度,因此降低催化活性。gydF4y2Ba如值取决于两个合成参数:pH值和热治疗。gydF4y2Ba然而,根据这些结果,光催化活性是喜欢当每个化合物的结构已经形成。gydF4y2Ba2,最好的活动4-dinitroaniline分解,是由锂hexatitanate溶胶-凝胶法形成晶体结构时,gydF4y2BaNagydF4y2Ba2gydF4y2Ba“透明国际”gydF4y2Ba6gydF4y2BaOgydF4y2Ba13gydF4y2Ba和gydF4y2BaKgydF4y2Ba2gydF4y2Ba“透明国际”gydF4y2Ba6gydF4y2BaOgydF4y2Ba13gydF4y2Ba由固态反应,gydF4y2Ba因此,它似乎是必须的晶体结构是一个重要的因素被认为是在光催化过程中。gydF4y2Ba在这些原子排列相似但同构的化合物gydF4y2BaTiOgydF4y2Ba6gydF4y2Ba正八面体变形gydF4y2Ba造成的碱性原子大小gydF4y2Ba是不同的gydF4y2Ba。gydF4y2Ba对这些实验扭曲的结构表现出越少gydF4y2Ba年代gydF4y2Ba光催化过程的主要活动。gydF4y2Ba降低铬的初步研究,gydF4y2BaNagydF4y2Ba2gydF4y2Ba“透明国际”gydF4y2Ba6gydF4y2BaOgydF4y2Ba13gydF4y2Ba溶胶-凝胶法是最好的催化剂。gydF4y2Ba 此外,合成方法中起着重要作用决定光催化材料的表面特征。gydF4y2Ba我gydF4y2Bat发现合成的固体通过溶胶-凝胶技术在基本条件下材料的催化行为的改善。gydF4y2Ba溶胶-凝胶技术使结构形成不同类型的缺陷的形成由于脱羟基在热处理过程。gydF4y2Ba该方法和合成条件的原因gydF4y2Ba不同的数量,类型和强度的表面活性gydF4y2Ba确定催化剂的催化性能。gydF4y2Ba 确认gydF4y2Ba我们通过格兰特承认联系提供的财政支持gydF4y2Ba35415 - u。gydF4y2Ba我们感谢Tessy洛佩兹博士她宝贵的溶胶-凝胶技术的监督。gydF4y2Ba我们也感谢Brenda费尔南德斯和露西亚卡斯蒂略执行初步铬(VI)光致还原作用实验。gydF4y2BaJ.C. Luna-UrzuagydF4y2Ba希望感谢联系颁发的奖学金。gydF4y2Ba 引用gydF4y2Ba1。gydF4y2Ba凯龙星,a . Fernandez-Alba罗德里格斯gydF4y2Ba和gydF4y2Bae . Garcia-Calvo“农药化学氧化:最先进的”。gydF4y2Ba,gydF4y2Ba窟。gydF4y2BaRes。gydF4y2Ba,gydF4y2Ba34gydF4y2Ba(2000)366 - 377。gydF4y2Ba 2。gydF4y2Baa . Fujishima t . n . Rao, d . a . Tryk二氧化钛光催化,j .光化学与光生物学C:光化学评论,gydF4y2Ba1gydF4y2Ba(2000)21。gydF4y2Ba 3所示。gydF4y2Baj . p . Wilcoxon“五氯苯酚的催化光致氧化使用半导体发光机制”,gydF4y2Ba期刊。化学。BgydF4y2Ba,gydF4y2Ba104年gydF4y2Ba(2000)gydF4y2Ba7334 - 7343。gydF4y2Ba 4所示。gydF4y2Baj·m·赫尔曼,“异质光催化:基础应用程序的各种类型的水污染物”,今天催化,gydF4y2Ba53gydF4y2Ba(1999)115 - 129。gydF4y2Ba 5。gydF4y2Ba井上y。,T. Kubokawa and K. Sato, “Photocatalytic Activity of Alkali-Metal Titanates Combined With Ru in the Decomposition of Water”,期刊。化学,gydF4y2Ba95年gydF4y2Ba(1991)4059 - 4063。gydF4y2Ba 6。gydF4y2Bac . Suckut r·a·豪伊a。r .西部和l . m . Torres-Martinez”的合成和性质gydF4y2BaHollandite-Like英航gydF4y2Ba3 xgydF4y2Ba李gydF4y2Ba(2 x + 4)gydF4y2Ba“透明国际”gydF4y2Ba(8-2x-Y)gydF4y2BaOgydF4y2Ba16gydF4y2Ba”,j .板牙。化学,gydF4y2Ba2gydF4y2Ba(1992)993 - 996。gydF4y2Ba 7所示。gydF4y2Bal . m . Torres-Martinez c . Suckut r·吉梅内斯和a·r·西”阶段形成和BaO-Li电气性能的系统gydF4y2Ba2gydF4y2BaO-TiOgydF4y2Ba2gydF4y2Ba”,j .板牙。化学,gydF4y2Ba4gydF4y2Ba(1994)5 - 8。gydF4y2Ba 8。gydF4y2Bac . Dusarrat r·a·豪伊c·马瑟·l·M。Torres-Martinez和a . r .西方”的晶体结构gydF4y2Ba英航gydF4y2Ba2gydF4y2Ba李gydF4y2Ba2/3gydF4y2Ba“透明国际”gydF4y2Ba16/3gydF4y2BaOgydF4y2Ba13gydF4y2Ba”,j .板牙。化学,gydF4y2Ba7gydF4y2Ba,gydF4y2Ba(1997)2103 - 2106。gydF4y2Ba 9。gydF4y2BaCh。k . Chan j·f·波特,y广,w .郭和陈志明Chan煆烧”效应在微观结构和纳米二氧化钛的光催化性能粉末由蒸汽水解”,gydF4y2Baj。陶瓷。Soc。gydF4y2Ba82年gydF4y2Ba(1999)566 - 573。gydF4y2Ba 10。gydF4y2Baa . Sclafani l·帕米萨诺和m . SchiavellogydF4y2Ba“TiO的制备方法的影响gydF4y2Ba2gydF4y2Ba的光催化降解苯酚在水分散”。gydF4y2Ba期刊。化学gydF4y2Ba。,gydF4y2Ba94年gydF4y2Ba(1990)829 - 832。gydF4y2Ba 11。gydF4y2Bak .年轻,Ching-BinHsieh。”gydF4y2Ba的光催化分解水TiO 4-DichlorophenolgydF4y2Ba2gydF4y2Ba悬浮体”,gydF4y2Ba水的研究gydF4y2Ba,gydF4y2Ba26gydF4y2Ba(gydF4y2Ba1992)1451 - 1456。gydF4y2Ba 12。gydF4y2Baj . Chen d F奥利。,W. H. Rulkens and H. Bruning, “Photocatalyzed Oxidation of Alcohols and Organochlorides in the Presence of Native TiO2gydF4y2Ba和镀金属TiOgydF4y2Ba2gydF4y2Ba悬浮液(第一部分)光催化活性和pH值的影响”,gydF4y2Ba窟。ResgydF4y2Ba。,gydF4y2Ba33gydF4y2Ba,gydF4y2Ba[3](1999)661 - 668。gydF4y2Ba 13。gydF4y2Bad·f·奥利w·H·j . Chen Rulkens和H。gydF4y2BaBruning”Photocatalyzed氧化醇类和Organochlorides本机TiO的存在gydF4y2Ba2gydF4y2Ba和镀金属TiOgydF4y2Ba2gydF4y2Ba悬浮液(第二部分)光催化机制”,gydF4y2Ba窟。ResgydF4y2Ba。,gydF4y2Ba33gydF4y2Ba[3](1999)669 - 675。gydF4y2Ba 14。gydF4y2Bac . j . Brinker R。Sehgal: k .拉曼普拉卡什和l . Delattre”gydF4y2Ba溶胶-凝胶法的策略控制孔隙度陶瓷材料:薄膜和散装”,亚博网站下载gydF4y2BaMat.Res。Soc。计算机协会。ProcgydF4y2Ba。,gydF4y2Ba368 (gydF4y2Ba1995)329 - 334。gydF4y2Ba 15。gydF4y2Bao . Novaro x Bokhimi, t·洛佩兹·e·桑切斯和r·戈麦斯gydF4y2Baj .板牙。ResgydF4y2Ba。gydF4y2Ba,gydF4y2Ba10gydF4y2Ba(1995)2788。gydF4y2Ba 16。gydF4y2Baz叮,g .问:卢和p·f·格林菲尔德”TiO的微晶阶段的作用gydF4y2Ba2gydF4y2Ba在多相光催化氧化苯酚在水里”,gydF4y2Ba期刊。化学gydF4y2Ba。B,gydF4y2Ba104年gydF4y2Ba(2000)gydF4y2Ba4815年。gydF4y2Ba 17所示。gydF4y2Ba诉Augugliaro l·帕米萨诺a . Sclafani c . Minero和e . Pellizeti毒理学和环境化学,16日,科学出版社,Inc .)、英国,(1988)89 - 109。亚博老虎机网登录gydF4y2Ba 18岁。gydF4y2Bal . j .太阳高,张问:“综合和比较高的光催化性质表面积金红石和锐钛矿二氧化钛纳米颗粒”,j .美国陶瓷协会、yabo214gydF4y2Ba86年gydF4y2Ba[10](2003)1677 - 1682。gydF4y2Ba 详细联系方式gydF4y2Ba |